SAFT-VR-Mie¶
The SAFT-VR-Mie EOS of Lafitte et al. (https://doi.org/10.1063/1.4819786) is based on the use of a Mie potential of the form
with
which allows for a better representation of thermodynamic properties in general, but not always.
[1]:
import teqp
teqp.__version__
[1]:
'0.19.1'
[2]:
import numpy as np
import pandas
import matplotlib.pyplot as plt
import CoolProp.CoolProp as CP
import scipy.integrate
[3]:
# Show two ways to instantiate a SAFT-VR-Mie model, the
# first by providing the coefficients, and the second
# by providing the name of the species. Only a very small
# number of molecules are provided for testing, you should
# plan on providing your own parameters.
#
# Show that both give the same result for the residual pressure
z = np.array([1.0])
model = teqp.make_model({
"kind": 'SAFT-VR-Mie',
"model": {
"coeffs": [{
"name": "Ethane",
"BibTeXKey": "Lafitte",
"m": 1.4373,
"epsilon_over_k": 206.12, # [K]
"sigma_m": 3.7257e-10,
"lambda_r": 12.4,
"lambda_a": 6.0
}]
}
})
display(model.get_Ar01(300, 300, z))
model = teqp.make_model({
"kind": 'SAFT-VR-Mie',
"model": {
"names": ["Ethane"]
}
})
display(model.get_Ar01(300, 300, z))
-0.04926724350863724
-0.04926724350863724
[4]:
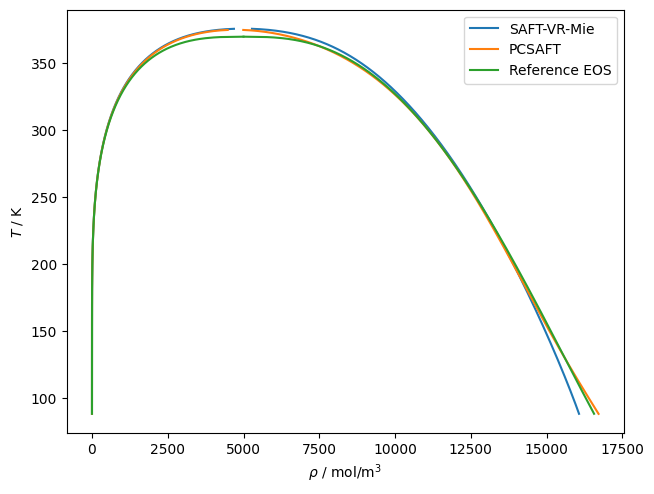
# Here is an example of using teqp to trace VLE for propane
# with the default parameters of PC-SAFT and SAFT-VR-Mie
# models
for kind in ['SAFT-VR-Mie', 'PCSAFT']:
j = {
"kind": kind,
"model": {
"names": ["Propane"]
}
}
model = teqp.make_model(j)
z = np.array([1.0])
Tc, rhoc = model.solve_pure_critical(300, 10000)
# Extrapolate away from the critical point
Ti = Tc*0.9997
rhoL, rhoV = model.extrapolate_from_critical(Tc, rhoc, Ti)
o = []
T = Ti
while T > 88:
rhoL, rhoV = model.pure_VLE_T(T, rhoL, rhoV, 10)
T -= 0.1
o.append({'rhoL': rhoL, 'rhoV': rhoV, 'T': T})
df = pandas.DataFrame(o)
line, = plt.plot(df['rhoL'], df['T'], label=kind)
plt.plot(df['rhoV'], df['T'], color=line.get_color())
# From the reference EOS of Lemmon et al. via CoolProp
name = 'Propane'
Tc = CP.PropsSI(name, 'Tcrit')
Ts = np.linspace(88, Tc, 1000)
rhoL = CP.PropsSI('Dmolar','T',Ts,'Q',0,name)
rhoV = CP.PropsSI('Dmolar','T',Ts,'Q',1,name)
line, = plt.plot(rhoL, Ts, label='Reference EOS')
plt.plot(rhoV, Ts, line.get_color())
plt.gca().set(xlabel=r'$\rho$ / mol/m$^3$', ylabel=r'$T$ / K')
plt.legend()
plt.tight_layout(pad=0.2)
plt.savefig('SAFTVRMIE_PCSAFT.pdf')
plt.show()
[5]:
# Time calculation of critical points
for kind in ['SAFT-VR-Mie', 'PCSAFT']:
j = {
"kind": kind,
"model": {
"names": ["Propane"]
}
}
model = teqp.make_model(j)
z = np.array([1.0])
%timeit model.solve_pure_critical(300, 10000)
1.25 ms ± 6.94 µs per loop (mean ± std. dev. of 7 runs, 1,000 loops each)
215 µs ± 188 ns per loop (mean ± std. dev. of 7 runs, 1,000 loops each)
[6]:
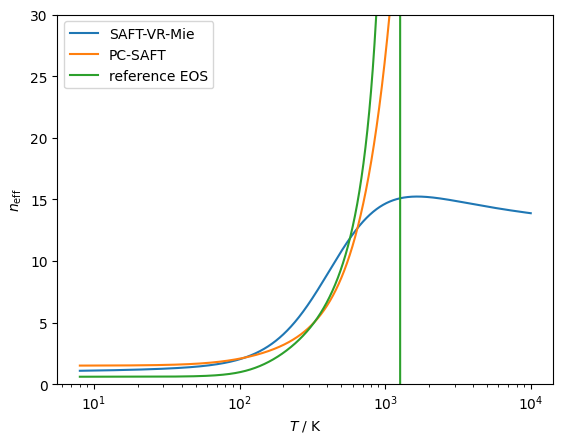
# Checking the effective hardness of interaction,
# the neff parameter defined in https://doi.org/10.1063/5.0007583
# SAFT-VR-Mie comes closest to the right behavior
modelVR = teqp.make_model({
"kind": 'SAFT-VR-Mie',
"model": { "names": ["Methane"] }
})
modelPCSAFT = teqp.make_model({
"kind": 'PCSAFT',
"model": { "names": ["Methane"] }
})
modelMF = teqp.build_multifluid_model(["Methane"], teqp.get_datapath())
for model, label in [(modelVR, 'SAFT-VR-Mie'),
(modelPCSAFT, 'PC-SAFT'),
(modelMF, 'reference EOS')]:
z = np.array([1.0])
rho = 1e-5
T = np.geomspace(8, 10000, 10000)
neff = []
for T_ in T:
neff.append(model.get_neff(T_, rho, z))
plt.plot(T, neff, label=label)
plt.xscale('log')
plt.ylim(0, 30)
plt.gca().set(xlabel=r'$T$ / K', ylabel=r'$n_{\rm eff}$')
plt.legend()
plt.show()
[7]:
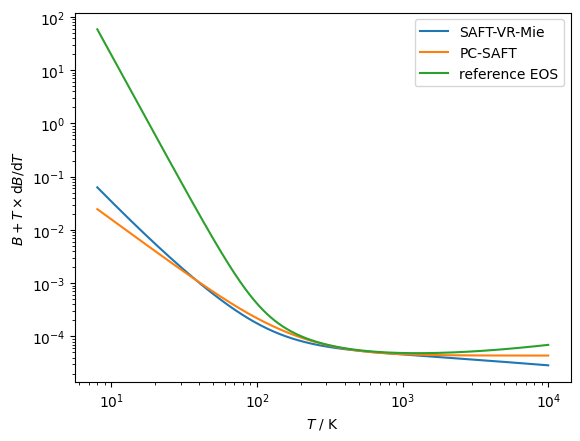
# Checking the temperature derivative of the virial coefficient
name = 'Methane'
modelVR = teqp.make_model({
"kind": 'SAFT-VR-Mie',
"model": { "names": [name] }
})
modelPCSAFT = teqp.make_model({
"kind": 'PCSAFT',
"model": { "names": [name] }
})
modelMF = teqp.build_multifluid_model([name], teqp.get_datapath())
for model, label in [(modelVR, 'SAFT-VR-Mie'),
(modelPCSAFT, 'PC-SAFT'),
(modelMF, 'reference EOS')]:
z = np.array([1.0])
T = np.geomspace(8, 10000, 10000)
n = 2
B, TdBdT, thetan = [],[],[]
for T_ in T:
TdBdT.append(model.get_dmBnvirdTm(n, 1, T_, z)*T_)
B.append(model.get_dmBnvirdTm(n, 0, T_, z))
thetan.append(B[-1]+TdBdT[-1])
plt.plot(T, thetan, label=label)
plt.xscale('log')
plt.yscale('log')
plt.gca().set(xlabel=r'$T$ / K', ylabel=r'$B+T\times$d$B$/d$T$')
plt.legend()
plt.show()
[8]:
# Time model instantiation
for kind in ['SAFT-VR-Mie', 'PCSAFT']:
j = {
"kind": kind,
"model": {
"names": ["Propane"]
}
}
%timeit teqp.make_model(j)
795 µs ± 2.5 µs per loop (mean ± std. dev. of 7 runs, 1,000 loops each)
331 µs ± 2.23 µs per loop (mean ± std. dev. of 7 runs, 1,000 loops each)
Calculation of diameter¶
The calculation of the diameter is based upon
but the integrand is basically constant from 0 to some cutoff value of \(r\), which we’ll call \(r_{\rm cut}\). So first we need to find the value of \(r_{\rm cut}\) that makes the integrand take its constant value, which is explained well in the paper from Aasen (https://github.com/ClapeyronThermo/Clapeyron.jl/issues/152#issuecomment-1480324192). Finding the cutoff value is obtained when
where EPS is the numerical precision of the floating point type. Taking the logs of both sides,
To get a starting value, it is first assumed that only the repulsive contribution contributes to the potential, yielding \(u^{\rm rep} = C\epsilon(\sigma/r)^{\lambda_r}\) which yields
and
Then we solve for the residual \(R(r)=0\), where \(R_0=\exp(-u/T)-EPS\). Equivalently we can write the residual in logarithmic terms as \(R=-u/T-\ln(EPS)\). This simplifies the rootfinding as you need \(R\), \(R'\) and \(R''\) to apply Halley’s method, which are themselves quite straightforward to obtain because \(R'=-u'/T\), \(R''=-u''/T\), where the primes are derivatives taken with respect to \(\sigma/r\).
[9]:
# Calculation of the residual function (needed for Halley's method)
import sympy as sy
kappa, j, lambda_r, lambda_a = sy.symbols('kappa, j, lambda_r, lambda_a')
u = kappa*(j**lambda_r - j**lambda_a)
display(sy.diff(u, j))
display(sy.simplify(sy.diff(u, j, 2)))
[10]:
# Here is a small example of using adaptive quadrature
# to obtain the quasi-exact value of d for ethane
# according to the pure-fluid parameters given in
# Lafitte et al.
epskB = 206.12 # [K]
sigma_m = 3.7257e-10 # [m]
lambda_r = 12.4
lambda_a = 6.0
C = lambda_r/(lambda_r-lambda_a)*(lambda_r/lambda_a)**(lambda_a/(lambda_r-lambda_a))
T = 300.0 # [K]
# The classical method based on adaptive quadrature
def integrand(r_m):
u = C*epskB*((sigma_m/r_m)**(lambda_r) - (sigma_m/r_m)**(lambda_a))
return 1.0 - np.exp(-u/T)
print('quasi-exact; (value, error estimate):')
exact, exact_error = scipy.integrate.quad(integrand, 0.0, sigma_m, epsrel=1e-16, epsabs=1e-16)
print(exact*1e10, exact_error*1e10)
j = {"kind": 'SAFT-VR-Mie', "model": {"names": ["Ethane"]}}
model = teqp.make_model(j)
d = model.get_core_calcs(T, -1, z)["dmat"][0][0]
print('teqp; (value, error from quasi-exact in %)')
print(d, abs(d/(exact*1e10)-1)*100)
quasi-exact; (value, error estimate):
3.597838592720949 3.228005612223332e-12
teqp; (value, error from quasi-exact in %)
3.597838640613809 1.331156429529301e-06