MATS Example using Experimental and Synthetic Data¶
Provided in the MATS v1.0 release are two examples using MATS in the Oxygen A-Band. The first uses experimental spectra and the second uses synthetic spectra. This overview steps through the common elements of both examples and highlights the differences between using experimental data and simulated spectra generated through the MATS.simulate_spectrum() function.
Import Modules and Set-Up¶
Both examples start with importing modules and setting up file locations
import numpy as np
import pandas as pd
import qgrid # only need if using the edit MATS.Edit_Fit_Param_Files class
import os, sys
import matplotlib.pyplot as plt
from matplotlib import gridspec
Append the system path to include the location of both the hapi and MATS modules
sys.path.append(r'C:\Users\Documents\MATS\MATS')# set location of HAPI.py module
from hapi import *
from MATS import *
Change the path to the working directory that contains experimental spectra or to the folder that you want to work in.
path = r'C:\Users\Documents\MATS\MATS\Examples\A-Band - Experimental Spectra'
os.chdir(path)
Load Spectra from files¶
There are two options for generating MATS.Spectrum objects. The first is from a file by instantiating an instance of the class. The second option is by using the MATS.simulate_spectrum() function described in the following section.
Before generating MATS.Spectrum objects from your experimental data, it is helpful to set some variables for terms that will be used in all of the MATS.Spectrum objects or throughout the fitting. In this example the minimum intensity threshold for simulation (IntensityThreshold), the minimum line intensity of lines fit in the analysis (Fit_Intensity), the order of the polynomial used in the baseline fits, and the names of columns used for the absorption, frequency, pressure, and temperature data are defined at the top of the example.
wave_range = 1.5 #range outside of experimental x-range to simulate
IntensityThreshold = 1e-30 #intensities must be above this value to be simulated
Fit_Intensity = 1e-24 #intensities must be above this value for the line to be fit
order_baseline_fit = 1
tau_column = 'Corrected Tau (us)' # Mean tau/us
freq_column = 'Total Frequency (Detuning)' # Total Frequency /MHz
pressure_column = 'Cavity Pressure /Torr'
temperature_column = 'Cavity Temperature Side 2 /C'
After that, 4 instances of the MATS.Spectrum class are instantiated from 4 experimental spectra. In the class instantiation, the mole fraction of the oxygen sample used is defined, the etalon amplitude and period are defined, the sample is confirmed to be at natural abundance, the diluent is set to air, and the columns defined for pressure, temperature, frequency, and absorbance data are set.
spec_1 = Spectrum('190510_2per_43_forfit',
molefraction = { 7 :0.01949}, natural_abundance = True, diluent = 'air',
etalons = {1:[0.001172, 1.19574]},
input_freq = True, frequency_column = freq_column,
input_tau = True, tau_column = tau_column, tau_stats_column = None,
pressure_column = pressure_column, temperature_column = temperature_column,
nominal_temperature = 296, x_shift = 0.00)
spec_2 = Spectrum('190510_2per_55_forfit',
molefraction = { 7 : 0.01949}, natural_abundance = True, diluent = 'air',
etalons = {1:[0.001172, 1.19574]},
input_freq = True, frequency_column = freq_column,
input_tau = True, tau_column = tau_column, tau_stats_column = None,
pressure_column = pressure_column, temperature_column = temperature_column,
nominal_temperature = 296, x_shift = 0.00)
spec_3 = Spectrum('190513_2per_82_forfit',
molefraction = { 7 :0.01949}, natural_abundance = True, diluent = 'air',
etalons = {1:[0.001172, 1.19574]},
input_freq = True, frequency_column = freq_column,
input_tau = True, tau_column = tau_column, tau_stats_column = None,
pressure_column = pressure_column, temperature_column = temperature_column,
nominal_temperature = 296, x_shift = 0.00)
spec_4 = Spectrum('190514_2per_126_forfit',
molefraction = { 7 :0.01949}, natural_abundance = True, diluent = 'air',
etalons = {1:[0.001172, 1.19574]},
input_freq = True, frequency_column = freq_column,
input_tau = True, tau_column = tau_column, tau_stats_column = None,
pressure_column = pressure_column, temperature_column = temperature_column,
nominal_temperature = 296, x_shift = 0.00)
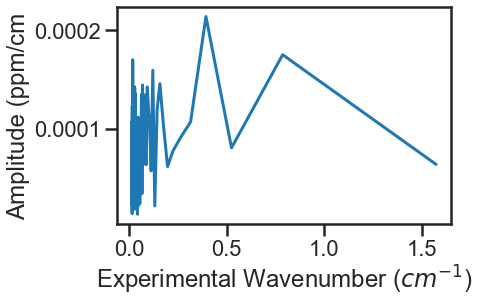
The MATS.Spectrum.plot_wave_alpha() function can be called to plot any of the spectra.
spec_1.plot_wave_alpha()
Simulate Spectra¶
If you are simulating spectra, opposed to reading them in from a file as discussed above, then you can use the MATS.simulate_spectrum() function.
When simulating spectra, the first step is to read in the reference line list. Generation of this line list is described in the Generating Parameter Line lists page. The following code reads in the reference line list as a pandas dataframe, then resets the working directory.
hapi = r'C:\Users\Documents\MATS\MATS\Linelists'
os.chdir(hapi)
PARAM_LINELIST = pd.read_csv('O2_ABand_Drouin_2017_linelist.csv')
path = r'C:\Users\Documents\MATS\MATS\Examples\A-Band - Synthetic Spectra'
os.chdir(path)
Just as you would do if reading in the experimental spectrum, this example defines some common simulation and fit variables. In addition to variables defined above, the minimum and maximum wavenumbers for the simulation and the simulation wavenumber spacing are defined. The baseline is defined by a polynomial where the array index is the parameter coefficient order, such that the [1, 0] would correspond to a linear baseline with a slope of 0 and an offset of 1.
wave_range = 1.5 #range outside of experimental x-range to simulate
IntensityThreshold = 1e-30 #intensities must be above this value to be simulated
Fit_Intensity = 1e-24 #intensities must be above this value for the line to be fit
order_baseline_fit = 1
sample_molefraction = {7 :0.002022}
wave_min = 13155 #cm-1
wave_max = 13157.5 #cm-1
wave_space = 0.005 #cm-1
baseline_terms = [0] #polynomial baseline coefficients where the index is equal to the coefficient order
The MATS.simulate_spectrum() function also allows for error to be added in the following ways:
to the absorption axis through signal-to-noise ratio (SNR). The SNR is implemented by adding gaussian noise to the spectra such that the (maximum alpha - minimum alpha) / noise is equal to the set SNR.
to the wavenumber axis through the wave_err parameter. The wavenumber error is implemented by adding a gaussian noise error of the specified magnitude to the wavenumber axis.
to the mole fraction through the molefraction_err parameter. The molefraction error is implemented as a percent error bias on each (could enter a negative percent error to get negative offset). This mimics the maximum impact that a constant error in sample mole fraction would have.
to the temperature/pressure through the temperature_err and pressure_err dictionaries. In experiments there are generally two type of errors with pressure and temperature measurements. The first is a constant bias in the reading. The second type of error is an actual change in the pressure/temperature during the collection of the spectrum. To account for both error types the pressure_err and temperature_err are dictionaries, where the keys correspond to ‘bias/per_bias’ (bias for temperature and per_bias for pressure), function (allows ‘linear’ or ‘sine’), and params. If the function is ‘linear’ then the param keys are ‘m’ and ‘b’ corresponding to the slope and interecept. If the function is ‘sine’ then the param keys are ‘amp’, ‘freq’, and ‘phase’ corresponding to the amplitude, period, and phase of the sine function. For both temperature and pressure, the pressure/temperature recorded in the simulated spectra output include the average pressure or temperature over the segment (analogous to the frequency of the pressure/temperature measurement in an experiment) and does not include the bias in pressure/temperature as this is would be an unknown in an experiment.
SNR = 10000
wave_error = 5e-5
temperature_err = {'bias': 0.01, 'function': None, 'params': {}}
pressure_err = {'per_bias': 0.01, 'function': None, 'params': {}}
molefraction_err = {7:0.01}
These parameters and the additional settings for filenames and number of segments can be used to call the MATS.simulate_spectrum() function setting the output equal to a variable as was done when the MATS.Spectrum was used. This makes it simple to transition code from analysis of experimental spectra to error analysis through simulations.
spec_1 = simulate_spectrum(PARAM_LINELIST, wave_min, wave_max, wave_space, wave_error = wave_error,
SNR = SNR, baseline_terms = baseline_terms, temperature = 25, temperature_err = temperature_err, pressure = 20,
pressure_err = pressure_err,
wing_cutoff = 50, wing_method = 'wing_cutoff', filename = '20_torr', molefraction = sample_molefraction, molefraction_err = molefraction_err,
natural_abundance = True, nominal_temperature = 296, IntensityThreshold = 1e-30, num_segments = 1)
spec_2 = simulate_spectrum(PARAM_LINELIST, wave_min, wave_max, wave_space, wave_error = wave_error,
SNR = SNR, baseline_terms = baseline_terms, temperature = 25, temperature_err = temperature_err, pressure = 40,
pressure_err = pressure_err,
wing_cutoff = 50, wing_method = 'wing_cutoff', filename = '40_torr', molefraction = sample_molefraction, molefraction_err = molefraction_err,
natural_abundance = True, nominal_temperature = 296, IntensityThreshold = 1e-30, num_segments = 1)
spec_3 = simulate_spectrum(PARAM_LINELIST, wave_min, wave_max, wave_space, wave_error = wave_error,
SNR = SNR, baseline_terms = baseline_terms, temperature = 25, temperature_err = temperature_err, pressure = 60,
pressure_err = pressure_err,
wing_cutoff = 50, wing_method = 'wing_cutoff', filename = '60_torr', molefraction = sample_molefraction, molefraction_err = molefraction_err,
natural_abundance = True, nominal_temperature = 296, IntensityThreshold = 1e-30, num_segments = 1)
spec_4 = simulate_spectrum(PARAM_LINELIST, wave_min, wave_max, wave_space, wave_error = wave_error,
SNR = SNR, baseline_terms = baseline_terms, temperature = 25, temperature_err = temperature_err, pressure = 80,
pressure_err = pressure_err,
wing_cutoff = 50, wing_method = 'wing_cutoff', filename = '80_torr', molefraction = sample_molefraction, molefraction_err = molefraction_err,
natural_abundance = True, nominal_temperature = 296, IntensityThreshold = 1e-30, num_segments = 1)
Generate a Dataset¶
The procedure for analysis for both spectrum class generation methods illustrated above is the same from this point on. The next step is to combine all desired MATS.Spectrum objects into a MATS.Dataset object, where we give the dataset a name. The order of polynomial to use for the baseline fit is also defined.
SPECTRA = Dataset([spec_1, spec_2, spec_3, spec_4], 'Line Intensity', baseline_order = order_baseline_fit)
The MATS.Dataset class contains a function to generate a baseline line list, analogous to the one for the parameter line list done outside of this example, based on the order of the baseline fit, etalons, molecules, x-shift parameters, and segments as defined by both the spectrum and dataset objects.
BASE_LINELIST = SPECTRA.generate_baseline_paramlist()
If the parameter line list hasn’t been read in from the .csv file, then do that now making sure to switch to the appropriate directories as needed.
hapi = r'C:\Users\Documents\MATS\MATS\Linelists'
os.chdir(hapi)
PARAM_LINELIST = pd.read_csv('O2_ABand_Drouin_2017_linelist.csv')
os.chdir(path)
Generate Fit Parameter Files¶
The next section of code uses the MATS.Generate_FitParam_File class to define what line shape to use for the initial fits, whether to use line mixing, the minimum line intensity to fit a line, mimimum intensity to included in the simulation, and for each line parameter whether that parameter is going to be constrained across all spectra or whether there will be a parameter for each spectrum (multi-spectrum vs single-spectrum fits) on a parameter by parameter basis. In the example below, the SDVP line profile without line mixing will be used to fit lines with line intensities greater than 1e-24 and the line centers and line intensities will be allowed to float for each line, while all other lines are constrained across all spectra in the dataset.
FITPARAMS = Generate_FitParam_File(SPECTRA, PARAM_LINELIST, BASE_LINELIST, lineprofile = 'SDVP', linemixing = False,
fit_intensity = Fit_Intensity, threshold_intensity = IntensityThreshold, sim_window = wave_range,
nu_constrain = False, sw_constrain = False, gamma0_constrain = True, delta0_constrain = True,
aw_constrain = True, as_constrain = True,
nuVC_constrain = True, eta_constrain =True, linemixing_constrain = True)
The next step is to generate fit parameter and baseline line lists that include columns that specify whether that parameter should be varied during fitting, in addition to adding error columns for the fit error for each parameter. For the following example the line centers, line intensities, collisional half-widths, and speed-dependent broadening terms will be floated for all main oxygen isotopes for lines where the line intensity is greater than 1e-24. Additionally, the baseline terms will float, as will the etalon amplitude and phase.
FITPARAMS.generate_fit_param_linelist_from_linelist(vary_nu = {7:{1:True, 2:False, 3:False}}, vary_sw = {7:{1:True, 2:False, 3:False}},
vary_gamma0 = {7:{1: True, 2:False, 3: False}, 1:{1:False}}, vary_n_gamma0 = {7:{1:True}},
vary_delta0 = {7:{1: False, 2:False, 3: False}, 1:{1:False}}, vary_n_delta0 = {7:{1:True}},
vary_aw = {7:{1: True, 2:False, 3: False}, 1:{1:False}}, vary_n_gamma2 = {7:{1:False}},
vary_as = {}, vary_n_delta2 = {7:{1:False}},
vary_nuVC = {7:{1:False}}, vary_n_nuVC = {7:{1:False}},
vary_eta = {}, vary_linemixing = {7:{1:False}})
FITPARAMS.generate_fit_baseline_linelist(vary_baseline = True, vary_molefraction = {7:False, 1:False}, vary_xshift = False,
vary_etalon_amp= True, vary_etalon_freq= False, vary_etalon_phase= True)
These functions will generate .csv files corresponding to these selections, which are read in by the MATS.Fit_DataSet class instantiation. This means that edits can be made manually to the .csv files or using the MATS.Edit_Fit_Param_Files class before the next code segment is run.
Fit Dataset¶
Instantiating the MATS.Fit_DataSet class reads in the information from the baseline and parameter linelists generated in the previous step. It also allows for limits to be placed on the parameters, so that they don’t result in divergent solutions. The example below includes several limits including limiting the line center to be within 0.1 cm-1 of the initial guess and the Line intensity to be within a factor of 2 of the intial guess. Placing limits on the parameters can be restrictive on the solution and cause the fit to not converge or return NaN for the standard error if it doesn’t allow for a local minima to be found.
fit_data = Fit_DataSet(SPECTRA,'Baseline_LineList', 'Parameter_LineList', minimum_parameter_fit_intensity = Fit_Intensity,
baseline_limit = False, baseline_limit_factor = 10,
molefraction_limit = True, molefraction_limit_factor = 1.1,
etalon_limit = False, etalon_limit_factor = 2, #phase is constrained to +/- 2pi,
x_shift_limit = True, x_shift_limit_magnitude = 0.5,
nu_limit = True, nu_limit_magnitude = 0.1,
sw_limit = True, sw_limit_factor = 2,
gamma0_limit = True, gamma0_limit_factor = 3, n_gamma0_limit= False, n_gamma0_limit_factor = 50,
delta0_limit = True, delta0_limit_factor = 2, n_delta0_limit = False, n_delta0_limit_factor = 50,
SD_gamma_limit = True, SD_gamma_limit_factor = 2, n_gamma2_limit = False, n_gamma2_limit_factor = 50,
SD_delta_limit = True, SD_delta_limit_factor = 50, n_delta2_limit = False, n_delta2_limit_factor = 50,
nuVC_limit = False, nuVC_limit_factor = 2, n_nuVC_limit = False, n_nuVC_limit_factor = 50,
eta_limit = True, eta_limit_factor = 50, linemixing_limit = False, linemixing_limit_factor = 50)
The next step is to generate the lmfit params dictionary object through the MATS.Fit_DataSet.generate_params() function. This translates baseline and parameter line list .csv files into the lmfit parameter dictionary that is used in the fits. After the parameters object is generated you can use the keys to set values and impose constraints on individual parameters, if desired. While this is not coded in the MATS toolkit, it is incredibly powerful as it lets you define min, max, vary, and expression values for any parameter. In the example below, two additional constraints are imposed on specific fit parameters. The first constrains all speed-dependent width parameters to be between the values of 0.01 and 0.25 and the second forces the amplitude of the etalon to be constant across all spectra.
params = fit_data.generate_params()
for param in params:
if 'SD_gamma' in param:
params[param].set(min = 0.01, max = 0.25)
if 'etalon_1_amp' in param:
if param != 'etalon_1_amp_1_1':
params[param].set(expr='etalon_1_amp_1_1')
The params file is then used to fit the spectra in the dataset using the MATS.Fit_DataSet.fit_data() function, where the result is a lmfit result object. The lmft prettyprint function prints the parameter fit results. Included below is an abbreviated prettyprint output that not only shows the fit result values and standard errors, but also highlights that constraints were imposed on the SD_gamma (speed dependent broadening) parameters and an expression was imposed on the etalon_amplitudes. It also shows that the there is a line intensity reported for every line (suffix number) and spectrum (after sw_) as the line intensities were not constrained to global fits. The reported sw shows that the fitted line intensity value is scaled by the minimum fit value. This aids in the fitting as line intensities are so much smaller than other values.
result = fit_data.fit_data(params, wing_cutoff = 25)
print (result.params.pretty_print())
Name Value Min Max Stderr Vary Expr Brute_Step
Pressure_1_1 0.07911 -inf inf 0 False None None
Pressure_2_1 0.06556 -inf inf 0 False None None
Pressure_3_1 0.04602 -inf inf 0 False None None
Pressure_4_1 0.02488 -inf inf 0 False None None
SD_delta_air_line_1 0 -inf inf 0 False None None
SD_delta_air_line_10 0 -inf inf 0 False None None
SD_delta_air_line_13 0 -inf inf 0 False None None
SD_delta_air_line_25 0 -inf inf 0 False None None
SD_delta_air_line_26 0 -inf inf 0 False None None
SD_gamma_air_line_1 0.1 0.01 0.25 0 False None None
SD_gamma_air_line_10 0.1137 0.01 0.25 0.0008273 True None None
SD_gamma_air_line_13 0.1313 0.01 0.25 0.001115 True None None
SD_gamma_air_line_25 0.09 0.01 0.25 0 False None None
SD_gamma_air_line_26 0.1 0.01 0.25 0 False None None
. . .
etalon_1_amp_1_1 0.001762 -inf inf 4.007e-05 True None None
etalon_1_amp_2_1 0.001762 -inf inf 4.007e-05 False etalon_1_amp_1_1 None
etalon_1_amp_3_1 0.001762 -inf inf 4.007e-05 False etalon_1_amp_1_1 None
etalon_1_amp_4_1 0.001762 -inf inf 4.007e-05 False etalon_1_amp_1_1 None
etalon_1_freq_1_1 1.196 -inf inf 0 False None None
etalon_1_freq_2_1 1.196 -inf inf 0 False None None
etalon_1_freq_3_1 1.196 -inf inf 0 False None None
etalon_1_freq_4_1 1.196 -inf inf 0 False None None
etalon_1_phase_1_1 -0.3479 -inf inf 0.04585 True None None
etalon_1_phase_2_1 -0.09384 -inf inf 0.04288 True None None
etalon_1_phase_3_1 -1.04 -inf inf 0.04446 True None None
etalon_1_phase_4_1 -1.266 -inf inf 0.04394 True None None
gamma0_air_line_1 0.04 -inf inf 0 False None None
gamma0_air_line_10 0.04501 -inf inf 4.919e-05 True None None
gamma0_air_line_13 0.04339 -inf inf 7.531e-05 True None None
gamma0_air_line_25 0.04 -inf inf 0 False None None
gamma0_air_line_26 0.04 -inf inf 0 False None None
. . .
sw_1_line_1 4.369 2.184 8.738 0 False None None
sw_1_line_10 4.735 2.4 9.598 0.0008558 True None None
sw_1_line_13 3.087 1.562 6.246 0.0007302 True None None
sw_1_line_25 2.083 1.042 4.166 0 False None None
sw_1_line_26 3.399 1.699 6.798 0 False None None
sw_2_line_1 4.369 2.184 8.738 0 False None None
sw_2_line_10 4.752 2.4 9.598 0.0006929 True None None
sw_2_line_13 3.091 1.562 6.246 0.0006913 True None None
sw_2_line_25 2.083 1.042 4.166 0 False None None
sw_2_line_26 3.399 1.699 6.798 0 False None None
sw_3_line_1 4.369 2.184 8.738 0 False None None
sw_3_line_10 4.744 2.4 9.598 0.0007446 True None None
sw_3_line_13 3.095 1.562 6.246 0.0007499 True None None
sw_3_line_25 2.083 1.042 4.166 0 False None None
sw_3_line_26 3.399 1.699 6.798 0 False None None
sw_4_line_1 4.369 2.184 8.738 0 False None None
sw_4_line_10 4.8 2.4 9.598 0.001158 True None None
sw_4_line_13 3.118 1.562 6.246 0.00117 True None None
sw_4_line_25 2.083 1.042 4.166 0 False None None
sw_4_line_26 3.399 1.699 6.798 0 False None None
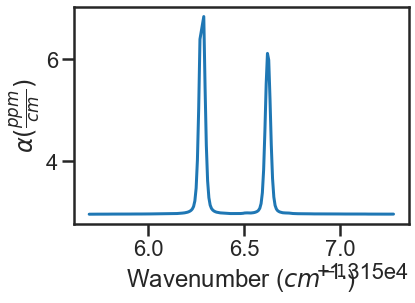
The last segment of code provides residual plots and updates residuals through the MATS.fit_data.residual_analysis() and MATS.Dataset.plot_model_residuals() functions, updates the parameter and baseline line lists through MATS.fit_data.update_params(), and generates a summary file with the fit results using MATS.Dataset.plot_model_residuals().
fit_data.residual_analysis(result, indv_resid_plot=True)
fit_data.update_params(result)
SPECTRA.generate_summary_file(save_file = True)
SPECTRA.plot_model_residuals()
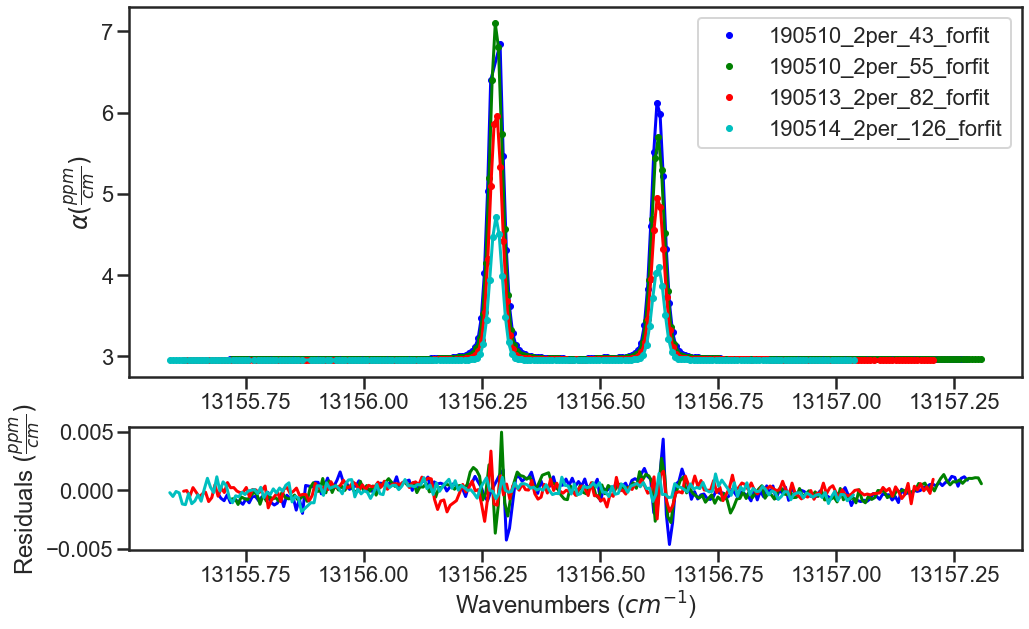
At the bottom of the experimental spectra example there is call to the MATS.Spectrum.fft_spectrum() function. If we hadn’t included the etalon, the fit residuals would look like this:
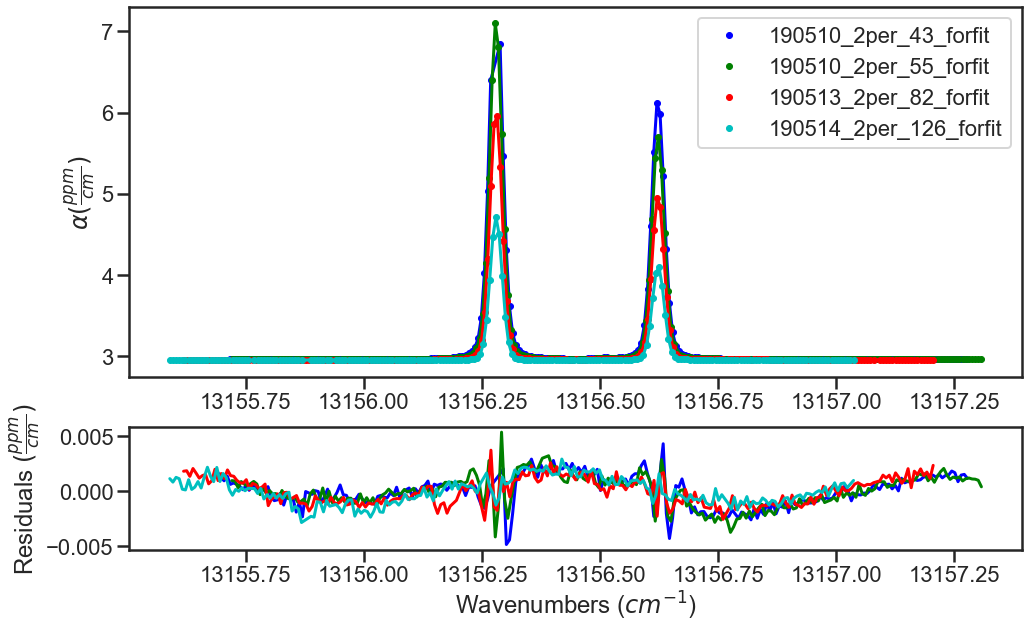
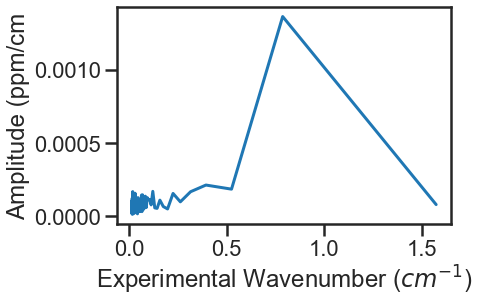
Calling the MATS.Spectrum.fft_spectrum() function on these residuals gives the following result with the most abundant period being 1.271443 cm-1 and an amplitude of 0.001364. The more etalon periods present in the spectral region being fit the more precise the etalon amplitude and frequency determined by the FFT will be.
Using these values as the etalon period and amplitude give the fit residuals shown above and when incorporated the FFT no longer shows a substantial peak at 1.271443 cm-1.